Syndrome de Sjögren primaire: du diagnostic au traitement.
Amara Pieren. Eva Benillouche. Service de Rhumatologie RHNe. |

En 1930, un ophtalmologue suédois examina une patiente souffrant d’arthrite et d’une extrême sécheresse des yeux et de la bouche. Il s’agissait du Dr. Henrik Sjögren qui donna non pas seulement le nom au Sd de Sjögren mais qui fut le premier à utiliser la tinction rose bengale pour étudier l’ampleur de l’atteinte cornéale, et qui établit le terme de “kérato-conjonctivite sèche”.
Le sd de Sjögren est une maladie multisystémique chronique auto-immune de progression lente et d’étiologie méconnue. Il se caractérise par une infiltration lymphocytaire des glandes exocrines et par la production d’autoanticorps. Il peut être primaire (pSS) ou être associé à d’autres maladies auto-immunes (Sd Sjögren secondaire). Il se manifeste typiquement par un syndrome sec sous forme de xérophtalmie et xérostomie accompagné de plaintes générales. Un patient sur trois va présenter une atteinte extra-glandulaire organo-spécifique.
Il s’agit de la deuxième maladie auto-immune la plus fréquente, après la polyarthrite rhumatoïde avec une prévalence estimée de 1–23 personnes par 10000 habitants dans les pays européens1 avec un pic d’incidence à l’âge de 50 ans. Le ratio femme/homme est de 9 :1.
Il est important de comprendre la maladie et d’apprendre à la reconnaitre. Il s’agit d’une pathologie qui peut être potentiellement grave et qui s’accompagne d’une forte morbidité et impact psycho-social.
2. Physiopathogénie
Les modèles physiopathologiques montrent une activation des cellules épithéliales des muqueuses, possiblement par une stimulation virale. Ceci induit une activation du système immun adaptatif et inné avec la sécrétion secondaire d’auto-anticorps. Ces auto-anticorps vont former des immunocomplexes qui maintiennent et amplifient la production d’interféron alpha, à l’origine des lésions tissulaires. La présence des centres ectopiques germinales dans les glandes salivaires surligne l’activation des cellules B, particularité caractéristique du pSS. 2
Le pSS se définit par une infiltration lymphocytaire des glandes lacrymales et salivaires avec une destruction tissulaire secondaire, à l’issue du syndrome sec. Cette infiltration peut également toucher l’épithélium d’autres organes donnant lieu aux complications systémiques, telles que la néphropathie interstitielle, la cholangite biliaire primitive auto-immune et la bronchiolite oblitérante. Le dépôt des immunocomplexes est responsable des manifestations extra-épithéliales comme le purpura palpable, la glomérulonéphrite associée aux cryoglobulines, l’atteinte interstitielle pulmonaire et la neuropathie périphérique. 3
3. Manifestations cliniques
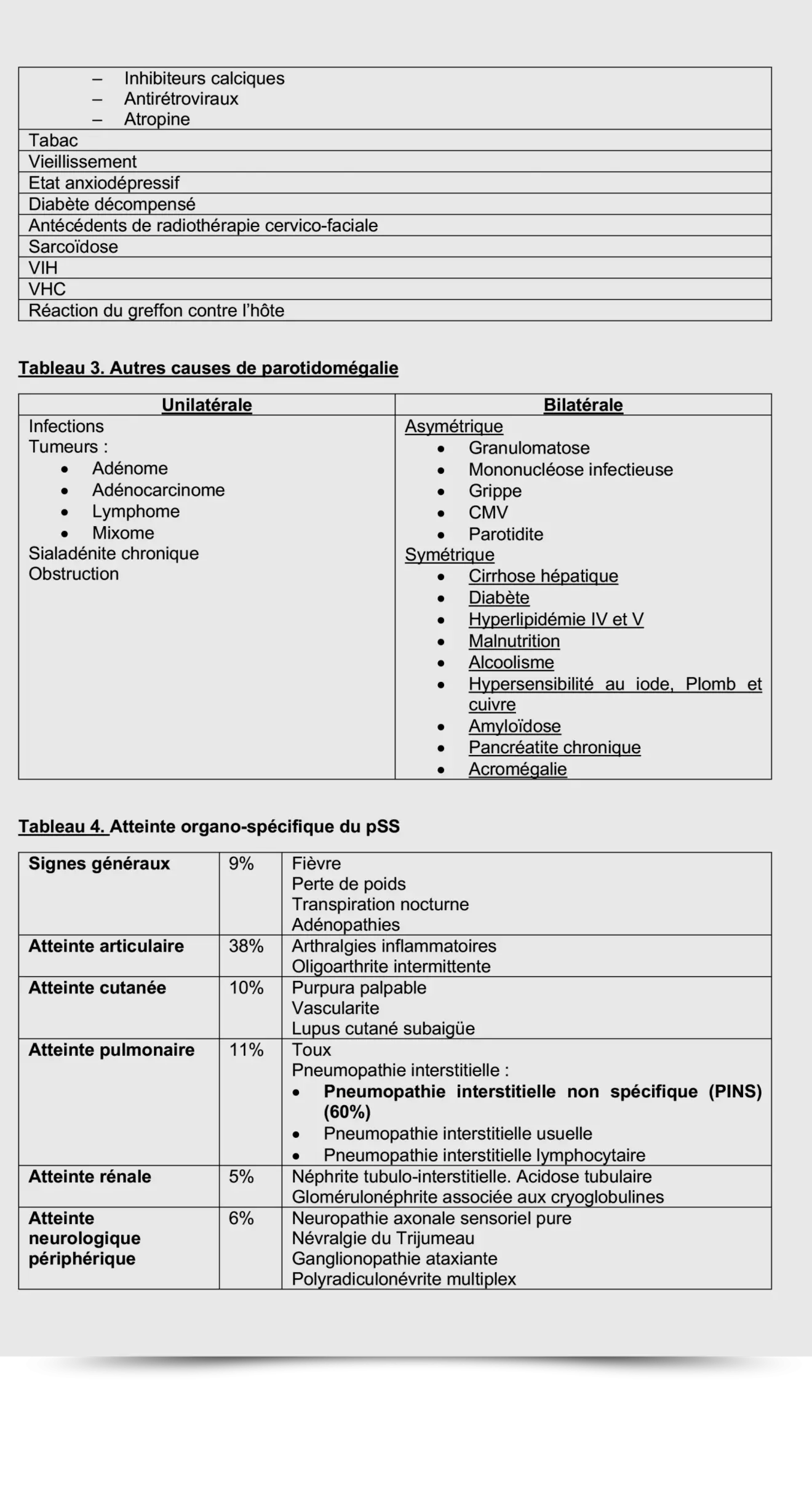
La triade clinique du pSS est composée d’un syndrome sec associé à une fatigue limitante et des arthralgies. Le challenge diagnostique repose sur la haute prévalence de ces symptômes dans la population générale. La sécheresse buccale et orale sont présentes dans 30% et 15% de la population générale respectivement, avec une prédominance du sexe féminin et une augmentation avec l’âge, d’où l’importance du diagnostic différentiel avec un impact majeur dans la prise en charge (Tableau 2. Causes syndrome sec).
L’atteinte des glandes exocrines se traduit par une kératoconjonctivite sèche (98%), une xérostomie (90%) et parfois, une tuméfaction des glandes salivaires (30-50%, voir tableau 3). La xérophtalmie se présente par une impression de corps étranger, fréquemment décrite comme du sable, ou d’une photophobie qui oblige la patient à instiller fréquemment des gouttes.4
L’hyposialorrhée se manifeste par une xérostomie pouvant s’associer à des brûlures buccales ou une dysphagie aux solides secs. Les patients boivent fréquemment pendant et en dehors des repas, y compris durant la nuit. La perte de l’effet tamponné de la salive et des larmes, induit des infections à répétition (caries récurrentes et ulcérations ou infections cornéennes). La sécheresse peut également s’exprimer au niveau cutanée, vaginale avec une dyspareunie secondaire ou bronchique, entrainant une toux sèche.
Les manifestations extra-glandulaires peuvent être le premier signe de la maladie, entrainant souvent un retard diagnostic du pSS. Devant le diagnostic d’une neuropathie périphérique ou pneumopathie interstitielle, entre autres, et en particulier s’il existe des symptômes de sécheresse oculaire ou orale, il est important de suspecter un pSS. Les atteintes organiques sont résumées dans le tableau 4.
Dans le pSS, le risque de développer un lymphome à cellule B est 15 à 20 fois plus élevé que dans la population générale (5-10% au cours de la vie)5, celui-ci étant secondaire à une hyperactivation de cellules B chronique. Il s’agit principalement de lymphomes non Hodgkinien à cellule B de bas grade. Les lymphomes apparaissent typiquement là où l’activité du pSS a été la plus agressive, comme les glandes salivaires (lymphomes associés aux muqueuses MALT). Les facteurs de mauvais pronostic vers une transformation lymphomatose sont résumés dans le tableau 5.
4. Diagnostic
Le diagnostic repose sur les éléments cliniques, l’objectivation du syndrome sec et les données biologiques et histologiques. Depuis 1967, plusieurs critères de classification ont été proposés par les différentes sociétés. Actuellement, les critères de classification ACR-EULAR 20166 sont les plus utilisés en pratique clinique (Tableau 6). Ceux-ci présentent comme nouveauté, la possibilité d’inclure des patients ayant des atteintes extra-glandulaires suspectes de pSS même sans syndrome sec. L’objectif est de diminuer le délai diagnostique et de faire connaitre aux praticiens ces formes moins fréquentes mais potentiellement plus graves.
A. Objectivation du syndrome sec
Le test de Schirmer est un test facile d’utilisation et de lecture en consultation. L’objectif est de mesurer la sécretion lacrimale. Il s’agit d’une bandelette rectangulaire de 5x35m , millimétrée, et placée dans le cul-de sac conjonctival inférieur de chaque œil près de l’angle externe. La longueur du papier humidifié par les larmes est relevée après cinq minutes. Le test est considéré pathologique en dessous de 5 mm / 5 minutes.
Une consultation ophtalmologique va nous permettre d’écarter des signes de kératoconjoctivite sèche avec les différents tests de coloration. L’érosion de la surface épithéliale oculaire est évaluée en instillant différents colorants : le vert de lissamine ou le rose Bengale pour la conjonctive et la fluorescéine pour la cornée. Il existe deux classifications permettant d’apprécier le degré d’atteinte oculaire en fonction de la distribution du marquage à la surface oculaire : l’« Ocular Staning Score » et le score de van Bijsterveld .
La mesure du flux salivaire permet de confirmer la présence d’une xérostomie. Elle peut être mesurée de manière stimulée ou non stimulée. Le flux salivaire total sans stimulation considéré normal est d’environ 0,4 ml/min.
B. Tests sérologiques
En cas de suspicion d’un pSS, nous allons rechercher la présence d’anticorps anti-nucléaire (ANA). Le premier screening sera fait par immunofluorescence sur les cellules Hep-2. En cas de confirmation de la présence des ANA et plus précisément, des ANA d’aspect moucheté, on recherchera les antinucléoprotéines (Anti-ENA) par ELISA ou Western-Blot (selon le laboratoire, d’autres test peuvent être utilisés), comprenant les spécificités anti-SSA / Ro 52/60kD et anti-SSB/La. Jusqu’à 30% des patients sont séronégatifs, ainsi, l’absence des ANA n’exclue pas la pathologie. La présence des AntiSSA/SSB est associée au risque de bloc congénital chez les nouveaux nés des patientes présentant ces anticorps. Ainsi, l’anti-RO52Kd est associé au risque de pneumopathie interstitielle. Malgré la haute prévalence des Anti-SSA/Ro et SSB/la dans le pSS, ceux-ci ne sont pas spécifiques de cette entité et peuvent apparaître dans d’autres maladies auto-immunes comme le lupus érythémateux diffus, les myopathies inflammatoires, certaines infections virales ou chez les sujets sains. De ce fait, le tableau clinique est primordial pour l’interprétation des auto-anticorps.
Le bilan immunologique comprendra également la recherche du facteur rhumatoïde, le complément et les cryoglobulines. Le dosage des immunoglobulines retrouve fréquemment une hypergammaglobulinémie avec une augmentation de la VS secondaire. Cet élément isolé peut nous orienter vers un pSS.
C. Biopsie des glandes salivaires
La biopsie des glandes salivaires a un rôle diagnostique et pronostique. Il s’agit d’un geste simple, mais non dépourvu de complications, qui est réalisé au niveau de la lèvre inférieure par une incision horizontale de 1 à 1,5 cm en regard de la muqueuse de la lèvre inférieure. L’objectif est de prélever au minimum 6 ou 7 glandes salivaires mineures.
L’histologie repose sur la mise en évidence d’infiltrats lymphocytaires dans la glande salivaire. Un focus (= foyer) correspond à un agrégat de ≥ 50 lymphocytes, caractéristique de la sialadénite lymphocytaire focale. La présence de ≥ 1 foyer par 4 mm2 de tissu glandulaire (grade ≥ 3 de Chisholm et Mason) est compatible avec un pSS.
Le Focus Score correspond au nombre de focus visibles sur une surface glandulaire de 4 mm2. Un Focus score ≥ 1 est considéré comme pathologique, suggestif de pSS. La recherche des focus score et des centres germinatifs est primordial, puisqu’il s’agit de signes de mauvais pronostic vers une transformation lymphomatose.
D. Imagerie des glandes salivaires
L’importance de l’échographie dans la prise en charge des pathologies inflammatoires ne peut pas être questionnée. Dans le pSS, l’ultrason est la technique d’imagerie par excellence, permettant d’évaluer le parenchyme glandulaire 7. Les foyers hypoéchogènes, correspondant aux zones inflammatoires qui caractérisent la maladie de Sjögren, sont recherchés au niveau d’une glande sous-mandibulaire et d’une glande parotide. Dans le cas des tuméfactions chroniques des parotides, l’échographie permet également d’écarter des signes suggestifs de lymphome. Toutefois, le PET est l’examen de référence en cas de suspicion de lymphome.
Plusieurs études ont suggéré une association entre l’échographie, les données histologiques et l’activité de la maladie. 8 Plus d’études sont nécessaires pour pouvoir établir le rôle de l’échographie dans la prise en charge du pSS.
Les méthodes de sialographie parotidienne et scintigraphie salivaire sont peu utilisées, étant donnée l’irradiation et leur manque de spécificité.
E. Evaluation de l’activité de la maladie et suivi
Il existe actuellement deux index validés pour évaluer l’activité de la maladie : le ESSPRI et ESSDAI. Le questionnaire ESSPRI est un questionnaire simple, faisable en consultation, (EULAR Sjögren’s Syndrome Patient Reported Index) qui s’intéresse à la sécheresse oculaire et orale, la fatigue et la douleur. Le ESSDAI par contre, est un index composé de 12 domaines, qui reprend les manifestations organiques.
La stratification du risque de lymphome est recommandée une fois tous les 1 ou 2 ans et doit comporter un hémogramme, une électrophorèse des protéines, le facteur rhumatoïde, C3 et C4 et les cryoglobulines.
5. Prise en charge du Syndrome de Sjögren
Le groupe EULAR a établi récemment les premières recommandations de prise en charge pharmacologique du pSS 9. Le traitement du pSS a malheureusement peu changé dans la dernière décennie. Il s’agit principalement d’un traitement symptomatique du syndrome sec. Divers immunosuppresseurs sont utilisés dans les atteintes organiques par parallélisme avec les autres maladies auto-immunes. Toutefois, on manque d’études de bonne qualité qui puissent démontrer l’efficacité et la sécurité des traitements employés.
La première ligne de traitement du syndrome sec repose sur l’utilisation des traitements topiques. La cyclosporine topique a démontré avoir un effet positif sur la qualité de la larme produite10. Les corticoïdes topiques ne sont pas recommandés en raison d’une efficacité limitée et des effets secondaires. Les agonistes muscariniques (pilocarpine et chlorhydrate de céviméline) ont prouvé leur efficacité dans le traitement de la xérostomie, et dans une moindre mesure, de la xerophtalmie 11.
Pour le traitement de la douleur, le traitement de première ligne reste l’acétaminophène. Il est important de faire la différence entre les arthralgies et l’inflammation articulaire ou arthrite, dont la prise en charge est drastiquement différente. L’introduction d’un traitement modulateur de la douleur doit être considéré. La gabapentine, prégabaline et duloxetine doivent être privilégiées, celles-ci induisant moins de sécheresse. L’hydroxychroloquine est employée dans les arthralgies inflammatoires récidivantes 12.
Dans l’atteinte extra-glandulaire, le choix du traitement doit être basé sur la sévérité de la maladie selon le ESSDAI. Les corticoïdes systémiques seront utilisés à la dose la plus faible et le moins de temps possible. Les traitements immunosuppresseurs (méthotrexate, léflunomide, azathioprine, mycophénolate mofétil et cyclophosphamide) sont introduits à but d’épargne cortisonique. De nos jours, aucune étude ne nous permet de choisir un traitement plutôt qu’un autre. Un traitement par rituximab doit être considéré dans les formes sévères et réfractaires 13. Le belimumab pourrait être essayé comme deuxième choix 14.
6. Conclusions
L’hétérogénéité clinique et biologique du syndrome de Sjögren rend le diagnostic difficile. Il est important de dépister les patients à risque de développer des atteintes extra-glandulaires sévères qui nécessitent une prise en charge rapide et multidisciplinaire. Un futur prometteur se dessine avec le développement de nouvelles thérapies.


Bibliographie :
- Brito-Zerón, P. et al. Sjögren syndrome. Nat. Rev. Dis. Primer 2, 1–20 (2016).
- Gottenberg, J.-E. et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s syndrome. Proc. Natl. Acad. Sci. U. S. A. 103, 2770–2775 (2006).
- Mariette, X. & Criswell, L. A. Primary Sjögren’s Syndrome. N. Engl. J. Med. 378, 931–939 (2018).
- Netgen. Syndrome de Sjögren : quand le suspecter et comment le confirmer ? Revue Médicale Suisse https://www.revmed.ch/RMS/2016/RMS-N-513/Syndrome-de-Sjoegren-quand-le-suspecter-et-comment-le-confirmer.
- Nocturne, G. & Mariette, X. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nat. Rev. Rheumatol. 9, 544–556 (2013).
- Shiboski, C. H. et al. 2016 ACR-EULAR Classification Criteria for primary Sjögren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol. Hoboken NJ 69, 35–45 (2017).
- Netgen. Syndrome de Sjögren : contribution de l’ORL au bilan diagnostique. Revue Médicale Suisse https://www.revmed.ch/RMS/2020/RMS-N-709/Syndrome-de-Sjoegren-contribution-de-l-ORL-au-bilan-diagnostique.
- Astorri, E. et al. Ultrasound of the salivary glands is a strong predictor of labial gland biopsy histopathology in patients with sicca symptoms. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 45, 450–454 (2016).
- Ramos-Casals, M. et al. EULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapies. Ann. Rheum. Dis. 79, 3–18 (2020).
- Ramos-Casals, M., Tzioufas, A. G., Stone, J. H., Sisó, A. & Bosch, X. Treatment of primary Sjögren syndrome: a systematic review. JAMA 304, 452–460 (2010).
- Vivino, F. B. et al. Pilocarpine tablets for the treatment of dry mouth and dry eye symptoms in patients with Sjögren syndrome: a randomized, placebo-controlled, fixed-dose, multicenter trial. P92-01 Study Group. Arch. Intern. Med. 159, 174–181 (1999).
- Gottenberg, J.-E. et al. Effects of hydroxychloroquine on symptomatic improvement in primary Sjögren syndrome: the JOQUER randomized clinical trial. JAMA 312, 249–258 (2014).
- Letaief, H. et al. Efficacy and safety of biological DMARDs modulating B cells in primary Sjögren’s syndrome: Systematic review and meta-analysis. Joint Bone Spine 85, 15–22 (2018).
- Mariette, X. et al. Efficacy and safety of belimumab in primary Sjögren’s syndrome: results of the BELISS open-label phase II study. Ann. Rheum. Dis. 74, 526–531 (2015).